npj Systems Biology and Applications, 2026
Signed, sealed, delivered: a generalizable model for living biotherapeutic dosing and metabolism
1 Department of Mathematics, Florida State University
2 Institute of Molecular Biophysics, Florida State University
3 Program in Neuroscience, Florida State University
Abstract
Living Biotherapeutic Products (LBPs) offer a promising therapeutic strategy for metabolic disorders rooted in gut microbiome dysfunction, yet quantitative frameworks for predicting their efficacy remain underdeveloped. We introduce the Bacterial Compartment Absorption and Transit (BCAT) model, a pharmacokinetic-pharmacodynamic framework that couples probiotic transit, endogenous microbiome metabolism, and enzymatic transformation within a unified dose-optimization setting. Building on the classical CAT model, BCAT incorporates mechanistically-derived colon compartments and treats dosing time as a control variable. We validate BCAT against clinical data for native choline metabolism and SYNB1618 probiotic trials, achieving 5% mean prediction error compared to ~30% for prior two-compartment models. Applying BCAT to trimethylaminuria (TMAU), we predict that ~109 CFU of engineered probiotic, administered 3–4 h before meals, achieves 95% reduction in systemic trimethylamine, matching healthy hepatic clearance. Global sensitivity analysis identifies enzyme expression level as the dominant design parameter, enforcing the broad applicability of this model. The BCAT framework generalizes to any gut microbiome-mediated metabolic disorder and provides quantitative dosing targets to guide live biotherapeutic development.
Introduction
Advances in synthetic biology have transformed the landscape of therapeutic development, enabling the engineering of living microorganisms as programmable drug delivery platforms. Bacteria such as Escherichia coli Nissle 1917, Lactococcus lactis, and Bacteroides ovatus have been engineered to express heterologous enzymes, secrete therapeutic peptides, and sense disease-associated biomarkers within the gastrointestinal tract. These Living Biotherapeutic Products (LBPs) exploit the gut as both a site of action and a route of systemic access, offering localized therapy with reduced off-target effects compared to conventional small-molecule drugs. Clinical programs have advanced LBPs targeting phenylketonuria, inflammatory bowel disease, hyperoxaluria, and Clostridioides difficile infection, with several candidates reaching Phase II trials.
Despite this progress, the development of LBPs has proceeded with remarkably limited quantitative support. Standard pharmacokinetic and pharmacodynamic (PK-PD) frameworks, designed for chemically defined drugs with predictable absorption and clearance, do not readily accommodate the biological complexity of living therapeutics: bacteria transit, replicate, express enzymes at variable rates, and interact competitively with the resident microbiome. Existing mathematical models of the gut microbiome have largely focused on ecological dynamics or metabolic flux analysis, without direct coupling to dosing decisions. The few compartmental models that have been proposed for LBPs—notably the two-compartment model of Charbonneau et al. and the three-compartment model of Lubkowicz et al.—simplify gastrointestinal anatomy to one or two well-mixed regions, omitting the colon entirely or treating it implicitly. This anatomical coarsening is consequential: for metabolites produced primarily by colonic bacteria, such as trimethylamine (TMA), the absence of explicit colon compartments introduces systematic prediction error.
In this paper, we present the Bacterial Compartment Absorption and Transit (BCAT) model, the first compartmental PK-PD framework that simultaneously couples probiotic transit, endogenous microbiome metabolism, and enzymatic transformation within a unified dose-optimization setting. BCAT builds on the classical Compartment Absorption and Transit (CAT) model, which discretizes the advection-absorption partial differential equation governing solute transport through the small intestine. The CAT framework models the concentration $\rho$ of a solute moving through a tube of length $L$ with velocity $v$ and absorption rate $K_a$:
$$\frac{\partial \rho}{\partial t} = -v \frac{\partial \rho}{\partial x} - K_a \rho$$Discretizing the spatial domain into $n$ compartments of equal length $\Delta x = L/n$ yields the system of ordinary differential equations:
$$\frac{d\rho_i}{dt} = K_t(\rho_{i-1} - \rho_i) - K_a \rho_i, \quad i = 1, \ldots, n$$where $K_t = v/\Delta x$ is the transit rate constant. The CAT model, with seven small intestinal compartments, has been validated extensively for oral drug absorption and forms the basis of the commercial GastroPlus and Simcyp platforms. The related ALT-CAT model of Mays and Nair extended this framework to include bacterial metabolism in the context of gut-derived uremic toxins, demonstrating the value of compartmental resolution for microbiome-mediated metabolite production.
BCAT extends the CAT architecture in several key directions. First, we derive the number of colon compartments and the colonic transit rate from radiopaque marker data, yielding a mechanistically grounded representation of large intestinal transit. Second, we introduce dynamic probiotic populations that transit alongside endogenous substrates and metabolites, with Michaelis-Menten kinetics governing enzymatic conversion. Third, we incorporate competitive interactions between probiotic enzymes and native microbiome metabolism, allowing the model to capture the therapeutic mechanism of substrate diversion. Finally, we treat dosing time—the interval between probiotic administration and meal consumption—as an explicit control variable, enabling optimization over both dose magnitude and timing.
We apply BCAT to trimethylaminuria (TMAU), a metabolic disorder arising from loss-of-function mutations in the hepatic enzyme flavin-containing monooxygenase 3 (FMO3). In healthy individuals, FMO3 oxidizes gut-derived TMA to the odorless trimethylamine $N$-oxide (TMAO) with approximately 95% efficiency. In TMAU patients, this clearance is impaired, leading to accumulation of volatile TMA in sweat, breath, and urine, producing a characteristic and socially debilitating fishy odor. Current management is limited to dietary restriction of choline-rich foods—an imprecise and burdensome intervention that does not address the underlying enzymatic deficit. An LBP expressing trimethylamine monooxygenase (TMM), the bacterial homolog of FMO3, could intercept TMA in the gut lumen before systemic absorption, effectively restoring the metabolic clearance absent in TMAU patients. BCAT provides the quantitative framework to determine the dose, timing, and enzymatic requirements of such a therapeutic.
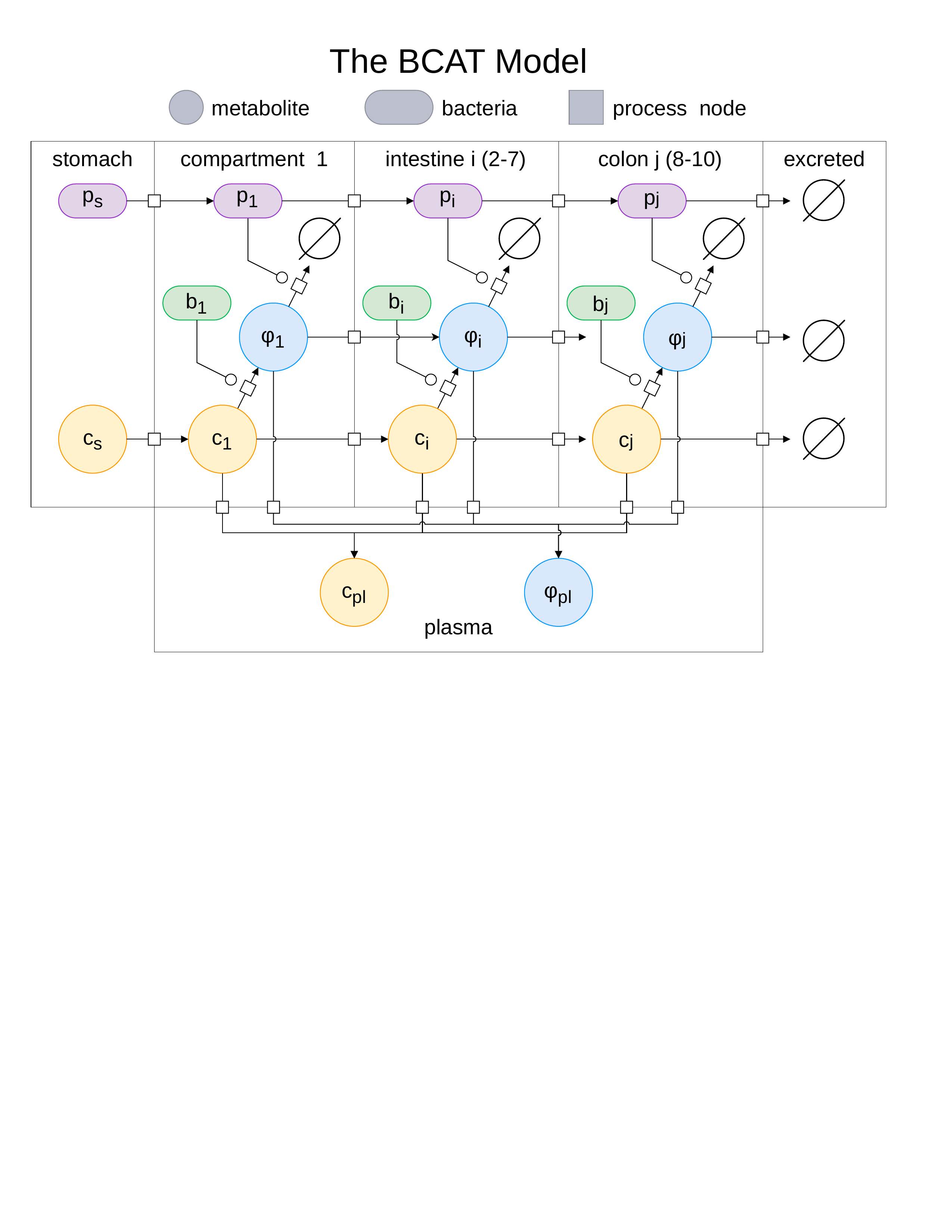
Results
Simulating trimethylaminuria
We define therapeutic efficacy through the ratio $\Gamma^*$ of cumulative TMA absorption into systemic circulation with probiotic treatment relative to the untreated case. Formally, $\Gamma^*$ represents the fraction of TMA that escapes enzymatic interception by the probiotic and reaches the bloodstream. The therapeutic objective is to identify the minimum probiotic dose $p_0$ that achieves $\Gamma^* = 0.05$, corresponding to a 95% reduction in systemic TMA exposure—the level of clearance provided by functional FMO3 in healthy individuals.
Baseline: no treatment
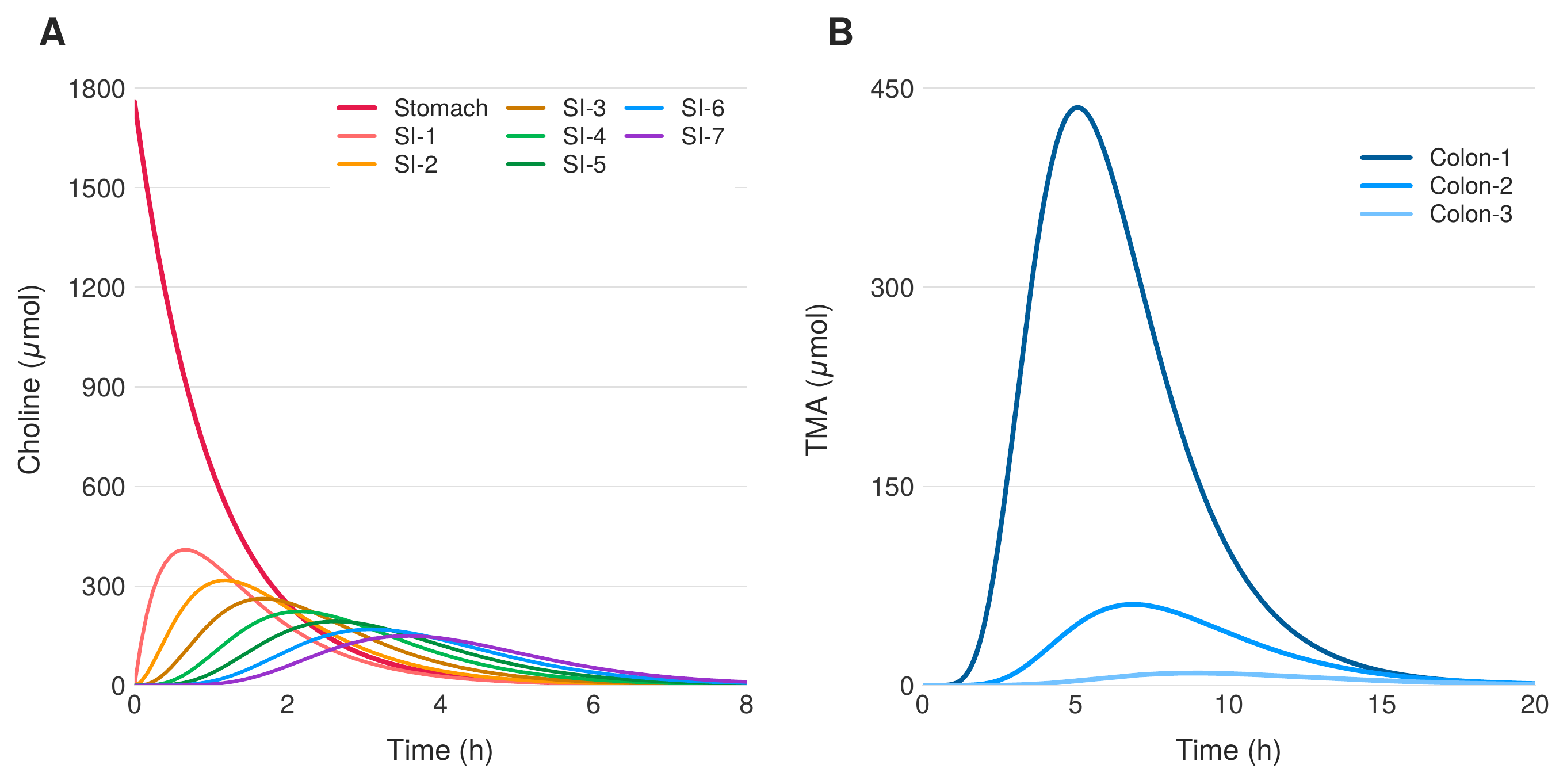
In the absence of probiotic intervention ($p_0 = 0$), dietary choline transits the gastrointestinal tract and undergoes unrestrained conversion to TMA by the endogenous microbiome. Figure 2 shows the temporal dynamics of choline and TMA concentrations across all gastrointestinal compartments following a standardized 1760 $\mu$mol choline bolus, representative of a high-choline meal. Choline transits rapidly through the stomach and small intestine, with partial absorption into the portal circulation. Upon reaching the colon, the resident microbiome converts the remaining choline to TMA via choline TMA-lyase activity. The solutions resemble a traveling wave with exponential decay, as each compartment receives flux from its predecessor while simultaneously losing material to absorption and forward transit. TMA production is dominated by the proximal colon, where bacterial density is highest, with concentrations peaking at approximately 440 $\mu$mol around 5 hours post-ingestion.
Probiotic treatment
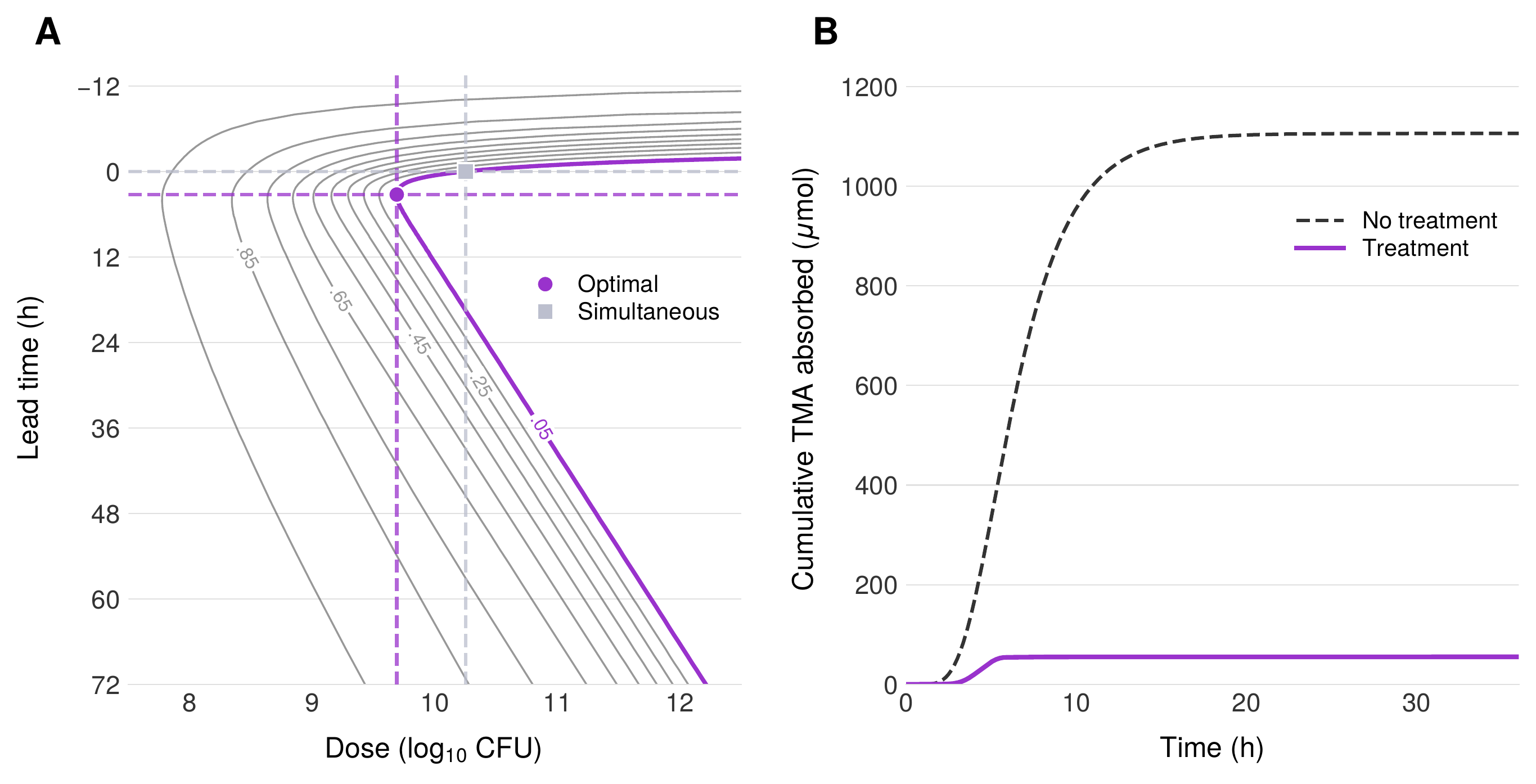
Introduction of a TMM-expressing probiotic diverts TMA away from systemic absorption by oxidizing it to TMAO within the gut lumen. BCAT predicts that approximately $10^{10}$ CFU achieves the therapeutic target of $\Gamma^* = 0.05$ when the probiotic is administered simultaneously with the choline-containing meal. This dose is well within the range of standard over-the-counter probiotic supplement capsules, which typically contain $10^9$–$10^{11}$ CFU. The model captures two distinct therapeutic goals: first, the direct reduction of systemic TMA to levels that eliminate the characteristic odor of TMAU; and second, the restoration of approximately 95% metabolic clearance, functionally matching the oxidative capacity of healthy hepatic FMO3.
Earlier probiotic administration
Administering the probiotic 3–4 hours before the choline-containing meal reduces the required dose by approximately four-fold compared to simultaneous administration. The mechanistic basis for this improvement is straightforward: probiotic bacteria administered in advance transit through the stomach and small intestine and accumulate in the colon before dietary choline arrives. When the choline bolus reaches the colonic compartments, a substantial probiotic population is already in place and actively expressing TMM, enabling immediate and efficient TMA interception. However, the benefit of early administration is not monotonic—administering the probiotic too far in advance is counterproductive, as the bacteria transit through and exit the colon before the substrate arrives. The optimal lead time of 3–4 hours reflects the balance between probiotic accumulation in the colon and the transit time of dietary choline from stomach to large intestine.
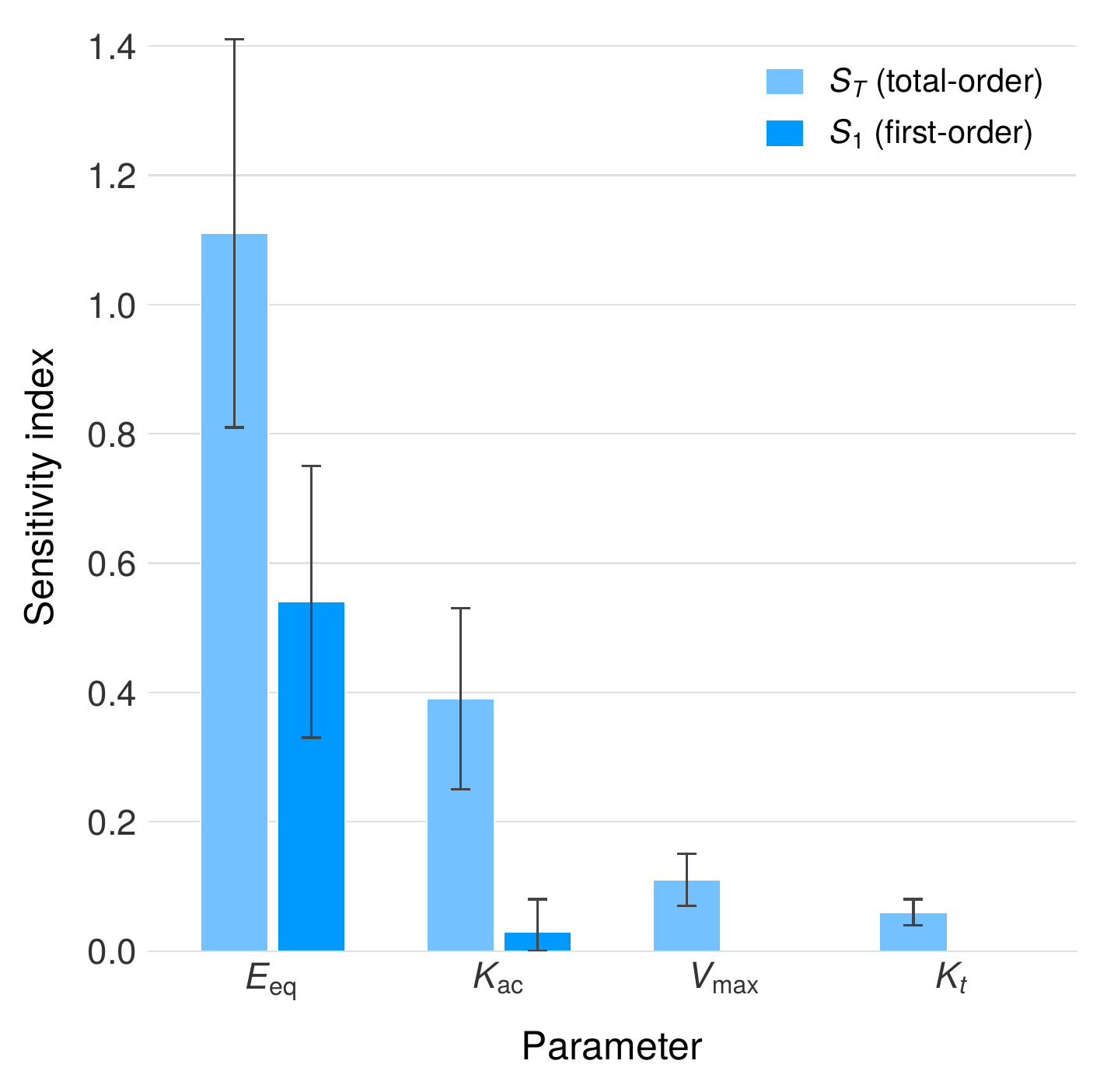
Global sensitivity analysis
To identify the parameters most influential in determining therapeutic outcome, we performed a Sobol variance-based global sensitivity analysis across the full BCAT parameter space. The enzyme expression level per cell, $E_{eq}$, emerges as the dominant design parameter with a first-order Sobol index of $S_1 = 0.54$ and a total-order index of $S_T = 1.11$, indicating that $E_{eq}$ accounts for over half of the output variance through its direct effect alone, with substantial additional contribution through interactions with other parameters. The disparity between $S_1$ and $S_T$ reveals significant interaction between enzyme expression level and the choline absorption rate $K_{ac}$, reflecting the competitive dynamics between host absorption and bacterial metabolism of the shared substrate.
These results carry three practical implications for LBP design. First, engineering effort should be concentrated on maximizing $E_{eq}$—the amount of active TMM enzyme expressed per bacterial cell—as this single parameter exerts the greatest leverage over therapeutic efficacy. Second, the relative insensitivity to physiological parameters such as gastric emptying rate and intestinal transit time indicates that BCAT predictions are robust to normal inter-individual variability in GI motility. Third, the low sensitivity to parameters that are difficult to measure in vivo, such as endogenous bacterial density, reduces the measurement burden required for patient-specific dose adjustment.
Discussion
The BCAT model represents the first compartmental PK-PD framework that simultaneously couples probiotic transit, endogenous microbiome metabolism, and enzymatic transformation within a single, optimizable architecture. By extending the classical CAT model with mechanistically derived colon compartments, dynamic probiotic populations, and competitive Michaelis-Menten kinetics, BCAT bridges the gap between ecological models of the gut microbiome and the dose-response frameworks required for rational therapeutic design. Validation against native choline metabolism data spanning three independent clinical studies and five decades of measurement (1951–1999) confirms the model's fidelity to endogenous physiology, while validation against SYNB1618 probiotic trial data demonstrates a mean prediction error of 5%, compared to approximately 30% for prior two-compartment approaches. The finding that 99.8% of TMA production occurs in the colon provides direct justification for the explicit colonic compartmentalization that distinguishes BCAT from earlier models.
Applied to TMAU, the model predicts that approximately $10^{9.7}$ CFU of a TMM-expressing probiotic, administered 3–4 hours before a choline-containing meal, achieves $\Gamma^* = 0.05$—a 95% reduction in systemic TMA that functionally restores healthy FMO3-level clearance. This dose falls within the range of commercially available probiotic formulations and could be delivered in a single capsule, suggesting clinical feasibility. The four-fold dose reduction achieved by optimizing administration timing demonstrates that dosing time is not merely a convenience parameter but a therapeutically meaningful control variable that should be explicitly optimized in LBP development. Global sensitivity analysis identifies enzyme expression level as the single most important engineering target, providing clear prioritization for synthetic biology efforts.
Several limitations of the current model should be acknowledged. BCAT does not incorporate probiotic growth, death, or colonization dynamics, treating the administered population as a fixed bolus that transits without replication or attrition. Endogenous bacterial densities are drawn from literature values rather than patient-specific measurements. Intracellular dynamics of enzyme expression—including transcriptional regulation, protein folding, and cofactor availability—are collapsed into the single parameter $E_{eq}$, which may not capture the full complexity of in vivo enzyme activity. These simplifications are appropriate for the acute, single-meal dosing scenario considered here but would need to be relaxed for chronic dosing regimens where colonization, washout, and adaptation become relevant.
Future extensions of the BCAT framework should incorporate population dynamics to model chronic dosing and steady-state colonization, enable patient-specific parameterization through Bayesian inference on individual GI transit measurements, and pursue experimental validation with TMM-expressing Bacillus subtilis or other GRAS chassis organisms. The compartmental architecture of BCAT is not specific to TMAU or TMA metabolism: any gut microbiome-mediated metabolic disorder in which a substrate is converted to a harmful metabolite by resident bacteria, and in which an engineered probiotic can intercept or divert that conversion, falls within the model's scope. Candidate applications include phenylketonuria, hyperoxaluria, and the production of gut-derived uremic toxins in chronic kidney disease.
Methods
Extending BCAT to the colon
The number of colon compartments $N$ and the colonic transit rate $K_{ct}$ were determined by fitting to radiopaque marker data from Metcalf et al., which tracked the spatial distribution of inert markers through the human colon via serial abdominal radiographs. We modeled colonic transit as a series of $N$ identical well-mixed compartments with uniform transit rate $K_{ct}$, analogous to the CAT discretization of the small intestine, and minimized the sum of squared errors (SSE) between predicted and observed marker distributions across all time points. The optimal fit was obtained with $N = 3$ compartments, corresponding to the ascending, transverse, and descending colon, with a transit rate of $K_{ct} = 3/35 \text{ h} = 0.086 \text{ h}^{-1}$. This three-compartment structure is consistent with the anatomical segmentation of the colon and the distinct transit characteristics of each region.
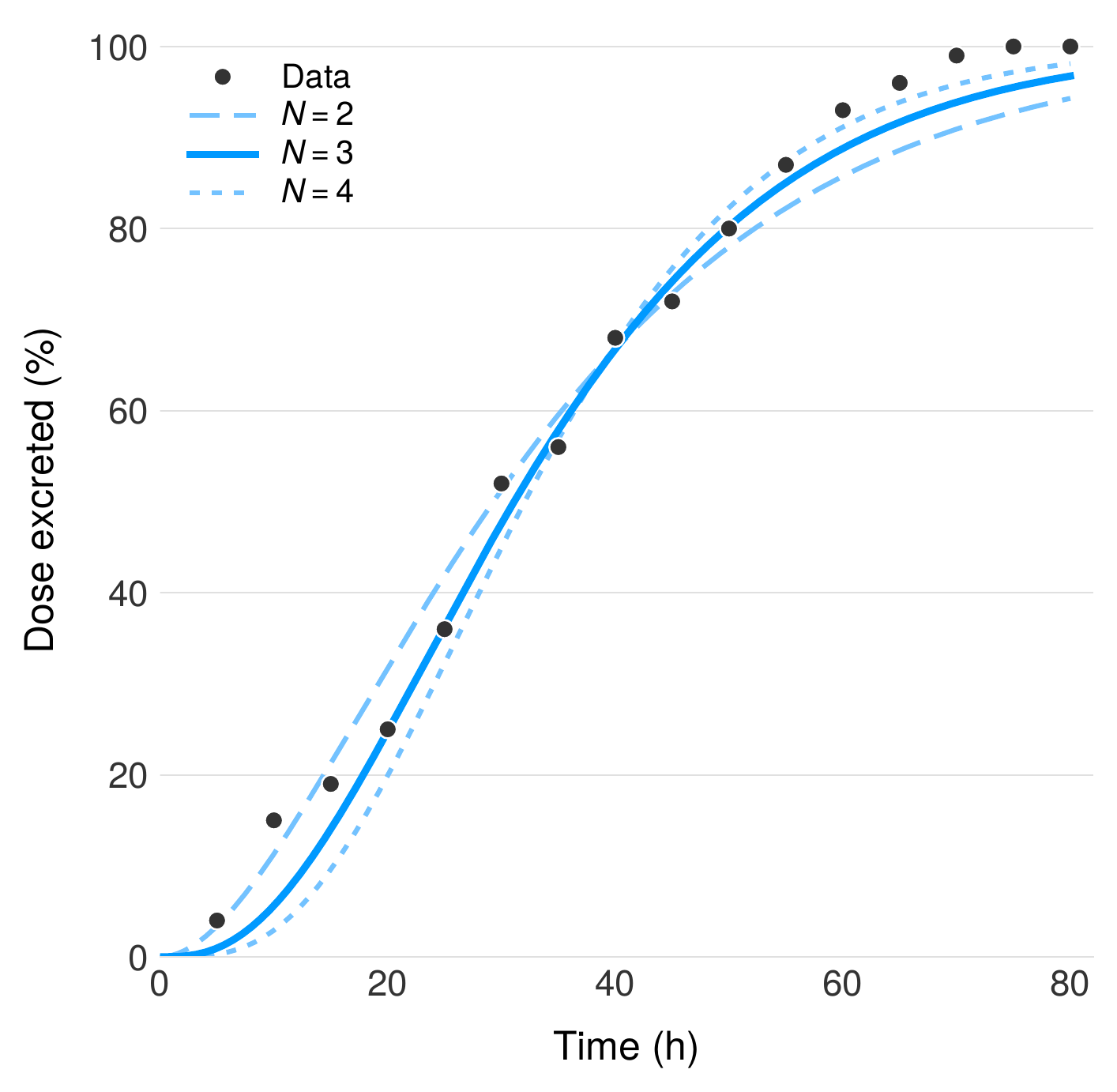
Validation against native choline metabolism
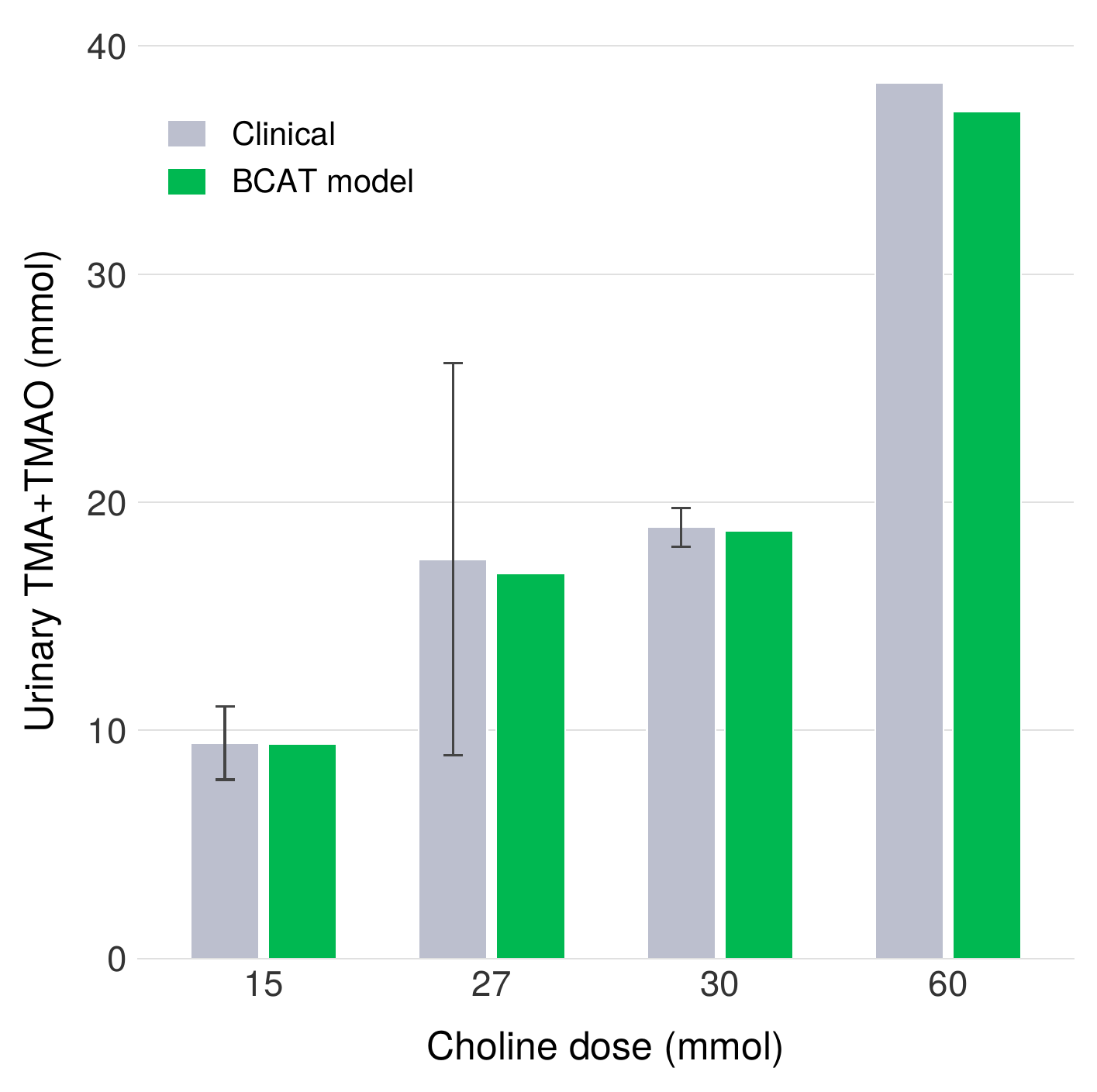
We validated the endogenous metabolism component of BCAT against three independent human studies spanning nearly five decades (1951–1999) that measured urinary TMA and TMAO excretion following oral choline administration. Across all three studies, the model reproduces the observed 63–65% bacterial conversion efficiency of dietary choline to TMA, a remarkably consistent finding given the diversity of experimental protocols, subject populations, and analytical methods employed. This agreement provides confidence that the microbiome metabolism parameters in BCAT reflect robust physiological constants rather than artifacts of any single dataset.
Validation against SYNB1618 clinical data
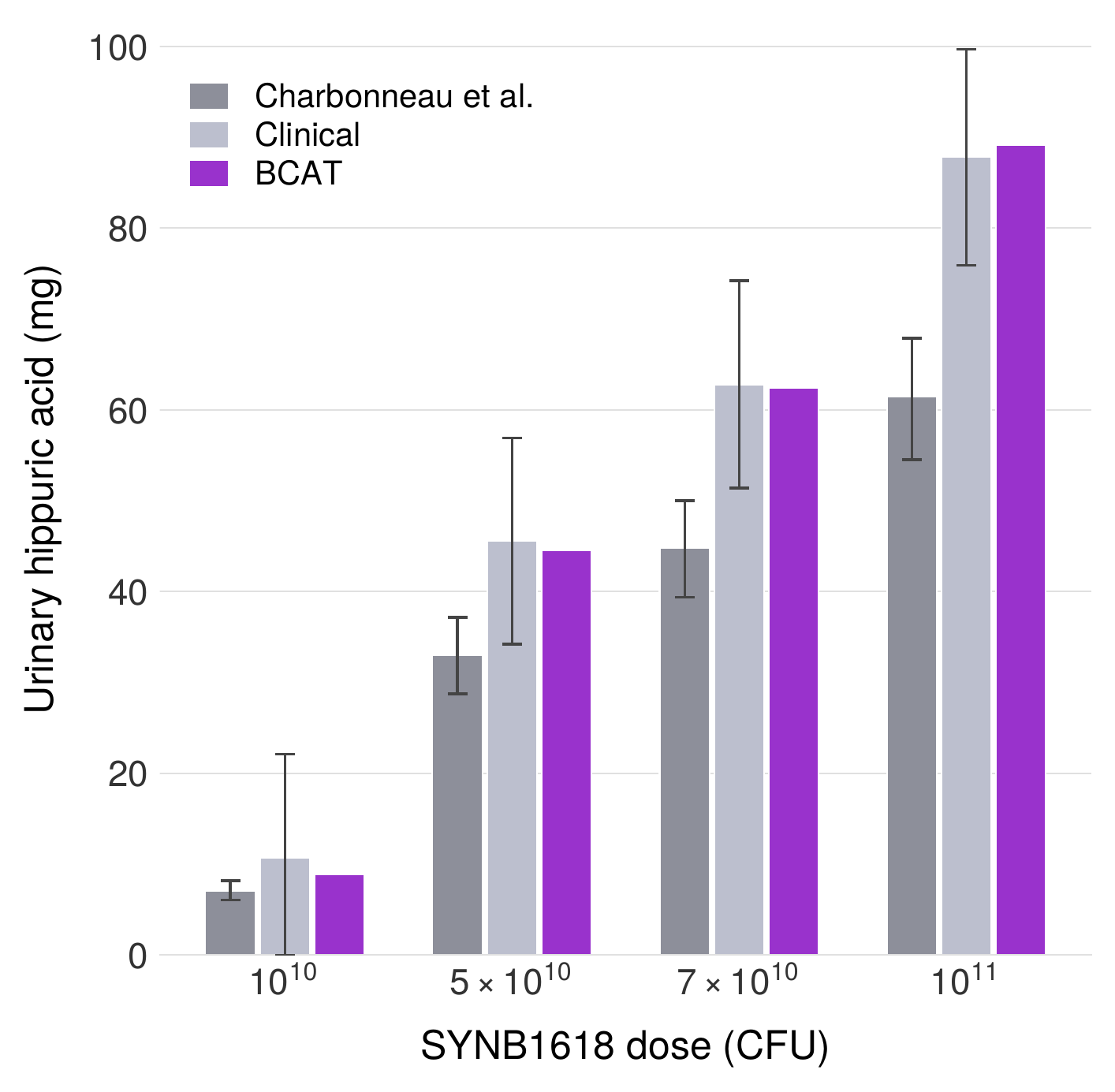
We further validated the probiotic component of BCAT against published clinical data from the SYNB1618 Phase I/IIa trial, in which an engineered E. coli Nissle 1917 strain expressing phenylalanine ammonia lyase was administered orally to healthy volunteers and patients with phenylketonuria. Using the full 10-compartment BCAT architecture (7 small intestinal + 3 colon compartments) with SYNB1618-specific kinetic parameters, the model achieves a mean prediction error of 5% against the reported urinary trans-cinnamic acid excretion data—a substantial improvement over the approximately 30% error reported for the two-compartment model of Charbonneau et al. applied to the same dataset. This validation demonstrates that the BCAT framework generalizes beyond TMA metabolism to other LBP applications.
Model Parameters
Table 1. Microbiome model parameters
| Parameter | Description | Value | Unit |
|---|---|---|---|
| $K_e$ | Gastric emptying rate | 0.984 | h−1 |
| $K_t$ | Small intestinal transit rate | 2.069 | h−1 |
| $K_{ct}$ | Colonic transit rate | 0.086 | h−1 |
| $K_{ac}$ | Choline absorption rate (SI) | 0.14 | h−1 |
| $K_{a\varphi}$ | TMA absorption rate (SI) | 0.744 | h−1 |
| $K_{a\varphi,co}$ | TMA absorption rate (colon) | 0.372 | h−1 |
| $V_{max}$ | Bacterial $V_{max}$ | 130 | fM h−1 (CFU/mL)−1 |
| $K_{mb}$ | Bacterial $K_m$ | 151 | $\mu$M |
Table 3. TMM kinetic parameters
| Parameter | Description | Value | Unit |
|---|---|---|---|
| $K_m$ | Michaelis constant (TMA) | 21.6 ± 1.9 | $\mu$M |
| $k_{cat}$ | Maximum reaction velocity | 1133.6 ± 58.6 | nmol·min−1·mg−1 |
| $E_{eq}$ | Enzyme per cell | 1.0 × 10−9 | mg/CFU |
This article is licensed under a Creative Commons Attribution 4.0 International License (CC-BY 4.0).